












城中活動
2026-08-18 10:00 上午
親親大自然|龜途生命教育體驗日
2026-08-14 2:00 下午
專題講座:癌症治療與口腔健康
2026-07-27 11:00 上午
插花藝術工作坊
疑難排解
我們有一群專業的醫護人員及相關朋友,隨時解答大家的疑難,立即提交疑問!
會員註冊
成為會員,可以第一時間接收由病患者和照顧者角度出發的資訊,立即行動!
或許你會想看
【港大醫學院】率先引進機械人輔助顯微外科手術
香港大學李嘉誠醫學院(港大醫學院)臨床醫學學院外科學系的臨床團隊,於香港率先引進機械人輔助顯微外科手術。團隊至 […]
中大成功拆解肝癌免疫治療耐藥性機制 揭一種免疫細胞具「除廢餵食」新功能助癌細胞耐藥性
免疫療法近年成為晚期肝癌的一線治療,惟大部分肝癌患者均對免疫療法呈耐藥性。醫學界多年來著力探究箇中耐藥機制,期 […]
三分鐘生成組織影像 支援術中即時診斷
香港科技大學(科大)研究團隊與科大培育的醫療科技初創公司遨天醫療科技有限公司(遨天醫療)成功研發出全球首創人工 […]
林順潮推動《ISOPT 癌病防控 香港宣言》在港正式啟動 倡議職場癌病防控新策略
6月14日,國際癌病防治醫學會(ISOPT)在香港正式啟動《ISOPT癌病防控香港宣言》(下稱《宣言》)。《宣 […]
中大與多名全球專家共同牽頭跨國肺癌研究 逾半晚期ALK陽性肺癌病人七年無惡化
中大與多名全球專家共同牽頭跨國肺癌研究 逾半晚期ALK陽性肺癌病人七年無惡化 因特定基因異常而引起的肺癌有望變 […]
【2026ASCO】Global Milestone in Cancer Care: Three Major Trends from the 2026 ASCO Annual Meeting Pancreatic Cancer Breakthrough and Precision Combination Therapies Take Center Stage
[June 2026 Comprehensive Report] The world’s largest on […]
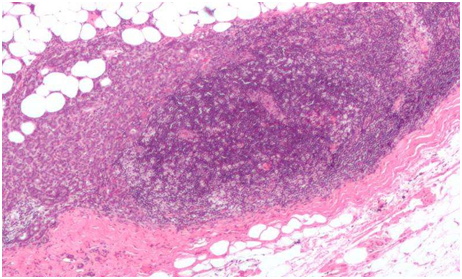
港大醫學院揭示癌症藥物抗藥性關鍵機制
2026年4月27日 香港大學李嘉誠醫學院(港大醫學院)生物醫學學院研究團隊發現,癌症病人對廣泛使用的癌症藥物 […]
港大醫學院中醫藥學院舉辦第13屆龐鼎元國際中醫藥研討會 匯聚本地海外專家推動中醫藥與中西醫結合
港大醫學院中醫藥學院舉辦第13屆龐鼎元國際中醫藥研討會 匯聚本地海外專家推動中醫藥與中西醫結合 香港大學李嘉誠 […]
中大研發經動脈碘栓塞化療無水新藥物配方治療肝細胞癌 無惡化存活期顯著延長一倍
中大研發經動脈碘栓塞化療無水新藥物配方治療肝細胞癌 無惡化存活期顯著延長一倍 肝癌位列本港第三大癌症殺手,亦是 […]
「2026 保誠健康論壇:優化患者醫療體驗」 薈聚醫療界領袖 共同支援香港及大灣區患者
「2026 保誠健康論壇:優化患者醫療體驗」 薈聚醫療界領袖 共同支援香港及大灣區患者 保誠保險 […]
【活動花絮】推動中西醫結合治療——乳癌研討會 圓滿落幕
由 癌症資訊網慈善基金 主辦的「推動中西醫結合治療 – 乳癌交流研討會」於今日 (2026年3月2 […]
【早期肺癌】術後輔助治療 減低復發風險
早期肺癌是有機會治癒,術後輔助治療減低復發風險 | 劉穎虹醫生 肺癌雖然係香港頭號癌症殺手,但早期肺癌是有機會 […]